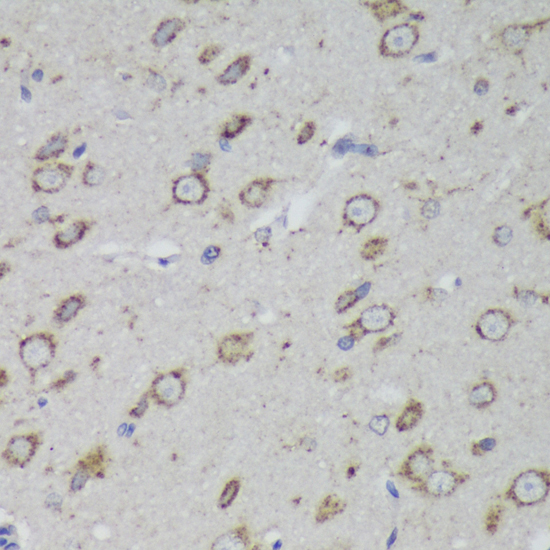
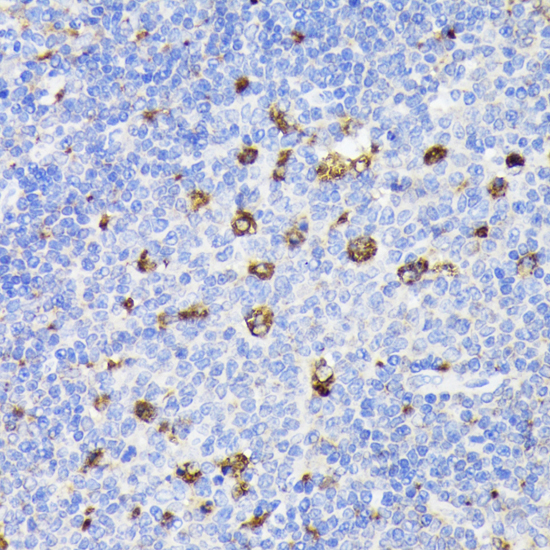
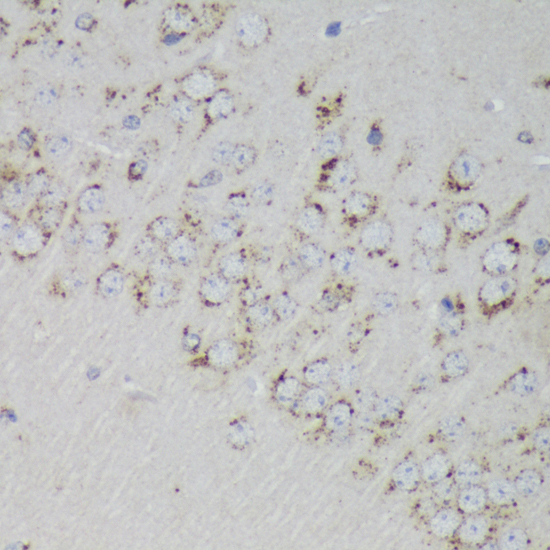
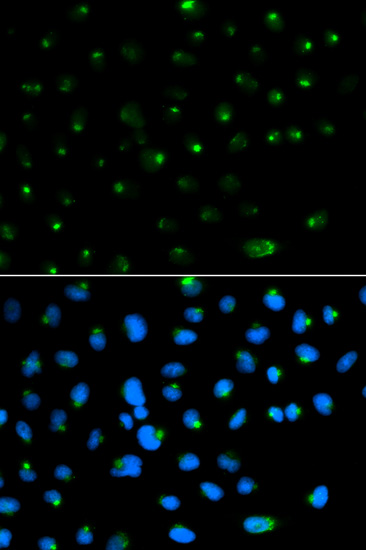
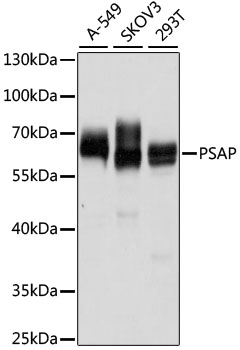
The PSAP gene encodes prosaposin, a precursor of four small nonenzymatic glycoproteins termed 'sphingolipid activator proteins' (SAPs) that assist in the lysosomal hydrolysis of sphingolipids. After proteolytic processing of the presaposin protein, these 4 released polypeptides are functional activators. Saposin A is encoded by residues 60 to 143 of PSAP, saposin B by 195 to 275, saposin C by 311 to 390, and saposin D by 405 to 487. They are four 12-14 kDa heatstable glycoproteins. Saposins A-D localize primarily to the lysosomal compartment where they facilitate the catabolism of glycosphingolipids with short oligosaccharide groups. Saposins A-D are required for the hydrolysis of certain sphingolipids by specific lysosomal hydrolases. (PMID: 2001789) Defects in PSAP are the cause of Gaucher disease, Tay-Sachs disease, and metachromatic leukodystrophy (PubMed: 2060627, PMID: 15773042). This PSAP antibody (10801-1-AP) is expected to recognize both saposin A and B.